block 21 (g2, g3, g4) stands as the third most representative of the overall n=75 population in this anabasis. jen just finished sectioning them, but some of the block 21 organoids, during the early sectioning out of 52 total slices (calculated from 26 slides x 2 slice/slide) have experienced what is known as the venetian blind artifact.
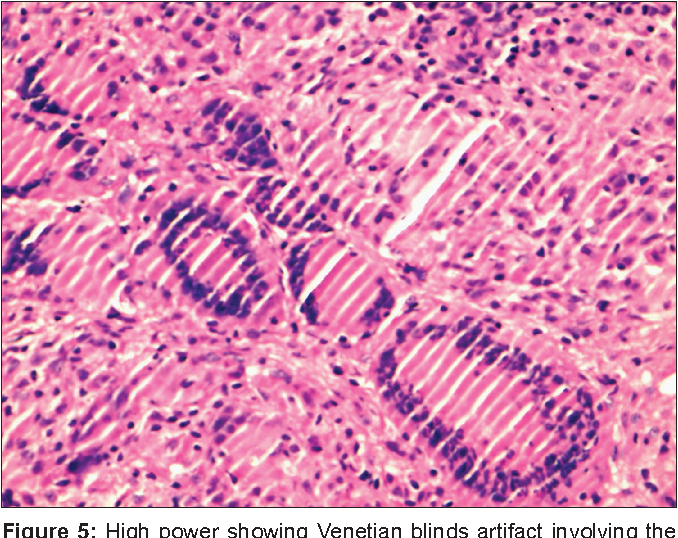
unfortunately, they are irreservable, and she wants to take just one test block that has the same OCT just without the actual organoids we care about to be used as a sacrificial lamb to find out the culprit behind all this. is it blade judder or temperature disequilibrium? she suggests it's the later. and it's only appropriate to find out what it is when the remaining 21 blocks (the other 4 are at histospring, with 3 remained as a block and 1 already sectioned and kept on unstained adhesion slides for IF already), alongside a testing block, are shipped to HistoSpring.
now, finding the culprit can save the rest of the population from potential disaster, but we have to know if block 21 is doomed for inclusion in the final dataset or not.
a few dependencies:
- i asked jen to tell me which organoid(s) in block 21, count of sections, the locational range affected by the venetian blind. she will tell me.
- on a more molecular level, directly know the clean vs. artifact distribution from beatriz when she does IF imaging
- understand the quality if excluding the artifact sections, which needs that distribution ratio.
- where within each affected section the artifact falls (1. within organoid cross section region or 2. the OCT gel aka background)
- how tolerative is the chosen quantification calculation for each respective protein biomarker expression (there's 6 total) toward the artifact.
- after every organoid's every biomarker's expression level is quantified, is g2 g3 and g4 — on an individual level — quantitatively outliers?
everything i just described above are human judgment to make a provisional default decision about inclusion or exclusion. default vs. secondary don't have to be ran separately, but rather in a single run of ENR training. so with these two possibilities considered, let's see how many total configurations, or the total amount of fittings we will proceed with ENR.
the full picture
fixed:
- Standardization: z-score per column (mandatory)
- Inner CV: 5-fold
- Outer CV: 5 randomized repeats × 5-fold
- Alpha grid: [0.1, 0.3, 0.5, 0.7, 0.9, 1.0] — 6 values
- Lambda: 100 auto-generated values per alpha
varies:
- biomarkers : CTIP2, TBR2, TBR1, HOPX, SOX2, MAP2 : count=6
- morphological feature subsets : {area}, {circ}, {ecc}, {area+circ}, {area+ecc}, {circ+ecc}, {all 3} : count=7
- temporal windows : D1-D47, D1-D46, D1-D45, ..., D1 : count=47
baseline configuration & fits:
6 x 7 x 47 = 1,974 model configurations
a configuration produces a bundle of target metrics (R², MAE, MAPE, etc.) w/ their mean ± std. For interpreting the std specifically, we will use Nadeau-Bengio corrected 95% CI. it has a hard rule on that the lower-bound CI value must > 0. that's it. we can optionally look into BH-FDR.
each config fits:
- 25 outer CV
- each outer train split runs 5 inner folds
- each inner fold evaluates 600 hyperparameter combinations (6 alphas x 100 lambdas)
[note: paper's α = sklearn l1_ratio, paper's λ = sklearn alpha]
25 x 5 x 600 = 75,000 fits per configuration.
baseline total: ~148 million fits.
with everything considered
the covariates
per window summary from back-extrapolation:
note: end day not listed has no covariates found
- D30: plate 3
- D22: plate 4
- D17: col 5, col 10, row C
- D16: row C
- D15: col 11
- D12: col 11
- D10: col 11
- D7: col 2
these are very scattered, and even when col 11 has shown up thrice, a good reason to, by default, exclude it from the covariate is that ENR prunes useless information. but we know that col 11's morphological outlier in some instances and only within the first epoch is upstream of what we are trying to have ENR to find: the correlation between morphology and biomarker expression. therefore, the default thing to do with covariates is to ignore any of them.
two combinations here regarding covariates:
- exclude col 11 as covariate (default)
- include col 11 as covariate (contingency)
venetian blind artifact in block 21
two combinations here, naturally:
- include g2,g3,g4
- exclude g2,g3,g4
notice that there's no default here, since we haven't seen all the metrics to make any assumption on whether the inclusion or exclusion is more reasonable. we have to evaluate everything in the "a few dependencies" section to make a final verdict before we actually build the ipynb.
grand total
a binary x a binary = 2x2 = 4 unique scenarios:
- exclude col 11 as covariate; include g2,g3,g4 (potentially default)
- exclude col 11 as covariate; exclude g2,g3,g4 (potentially default)
- include col 11 as covariate; include g2,g3,g4 (confirmed backup)
- include col 11 as covariate; exclude g2,g3,g4 (confirmed backup)
maximum model configurations: 1,974 x 4 = 7,896
fits per configuration remains unchanged: 75,000
maximum grand total fits: 7,896 x 75,000 = 592.2 million
let the model speaks for itself.